Viewing Job Results
When a job has completed, from the Job Manager page click either the job name or the View Job icon ( ).
).
Viewing Spectra
WebMO can display a variety of molecular spectra, depending on the capabilities of the underlying engine. Displayed IR and NMR spectra are interactive. Clicking on a peak in the spectrum will display the normal mode vibrations or atoms associated with the peak.
Examples are given below for but-1-en-2-ol.
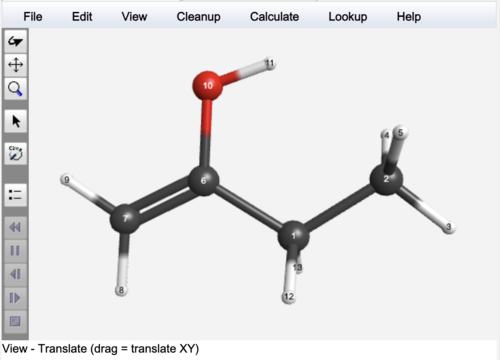
But-1-en-2-ol Optimized Geometry
Displaying Infrared and Raman Spectra
After a Vibrational Frequencies calculation, the normal mode frequencies are tabulated along with infrared intensities (and Raman and VCD intensities if available). The infrared, Raman, or VCD spectrum can be displayed by clicking the Spectrum, Raman, or VCD Spectrum view icon ().
Electronic structure calculations are well-known to systematically over-estimate vibrational frequencies. Thus WebMO automatically applies a Frequency Scale Factor to the computed frequencies for common methods and basis sets from the NIST table of precomputed vibrational scaling factors. (The unscaled frequency calculated by the computational engine is reported in parentheses.) The Frequency Scale Factor may also be set manually.
The peak width in the displayed spectrum may be set manually with the Peak Width value.
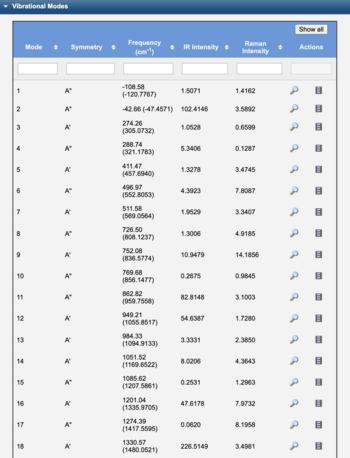 Vibrational Modes Table |
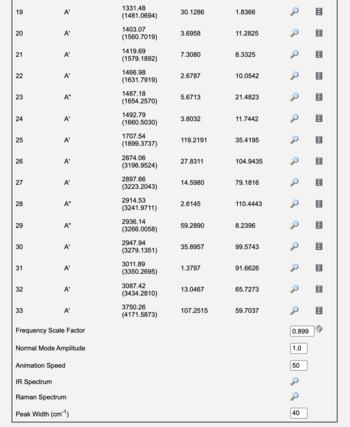 |
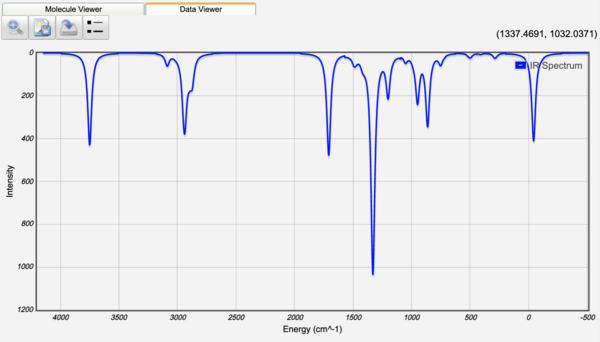
Infrared Spectra
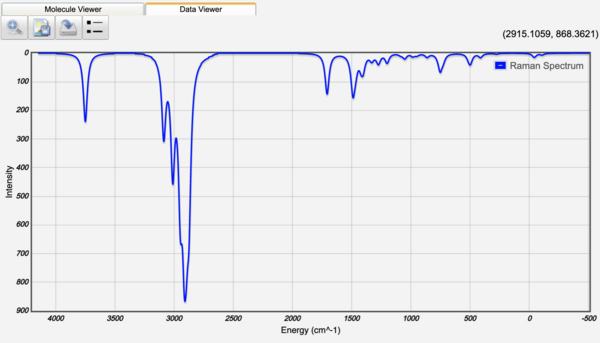
Raman Spectra
Clicking on a peak in the IR or Raman spectrum will display the normal mode vibration associated with the peak!
Displaying UV-Vis Spectra
After an Excited States Calculation, the excited state energies and transition intensities are tabulated. The UV-VIS spectrum can be displayed by clicking the UV-Vis Spectrum view icon (). Units can be converted between nm and eV. The peak width is set manually by the Peak Width value.
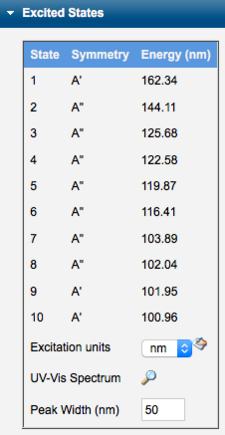
Excited States
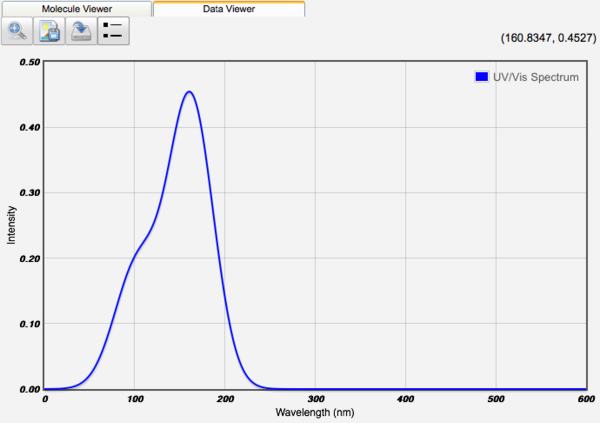
UV-Vis Spectra
Displaying NMR Spectra
After a NMR calculation, the chemical shifts of each atom are tabulated. 1H and 13C shifts are displayed relative to TMS values at the same basis and theory, if available. 1H and 13C NMR spectra can be displayed by clicking the View icon (). Relative frequencies and equal intensities are used. The peak width is set manually by the Peak Width value.
1H and 13C shifts can be shifted relative to a spectral feature by entering an offset value in the TMS shielding textbox and clicking the Shift NMR Spectrum icon ( ).
).
NMR-equivalent protons are identified for most molecules, and their chemical shifts are averaged. Proton splitting is the simulated using the specified magnetic field strength and coupling constant in order for 1H NMR spectra to look like experimental spectra. Since 13C NMR spectra are typically acquired as decoupled spectra, they are plotted without any splitting to look like experimental spectra.
If spin-spin coupling constants are calculated, then they are reported in a table and the 1H NMR spectrum uses them instead of the value specified by the user.

Chemical Shifts
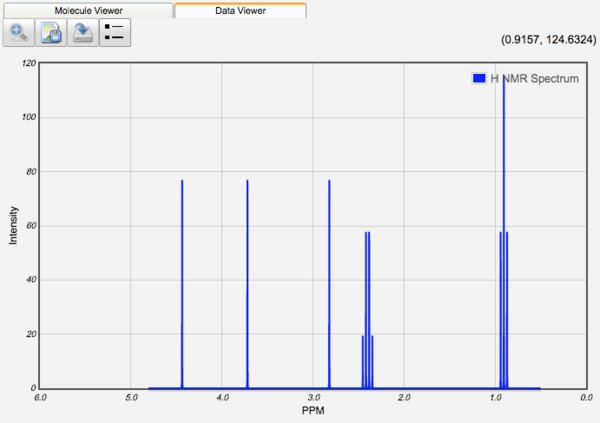
1H NMR Spectra
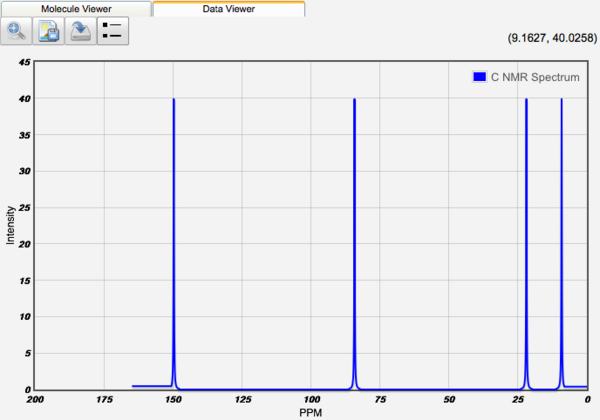
13C NMR Spectra
Clicking on a peak in the NMR spectrum will highlight the atoms responsible for the peak!
Displaying Vibrational Circular Dichroism (VCD) Spectra
After a Vibrational Frequencies Calculation for which Calculate VCD Intensities was selected on the Advanced Job Options tab, the VCD intensity of each vibrational mode is displayed in the results table. The VCD spectrum can be displayed by clicking the VCD Spectrum view icon (). This allows for the absolute determination of stereochemistry for a chemical compound.
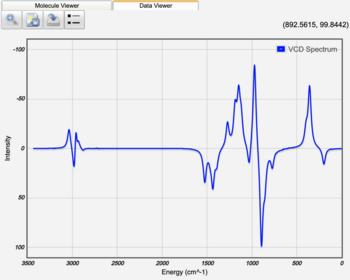 VCD Spectrum of R-Propylene Oxide |
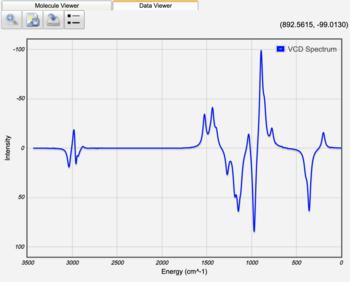 VCD Spectrum of S-Propylene Oxide |
Specifying Data Viewer Options
Spectra are displayed in the DataViewer tab of the View Results page. The following options exists for customizing or saving spectra:
Zoom in: Draw a box by clicking and dragging the mouse, and the spectrum will zoom to the selected area.
Reset zoom: Click the Reset Zoom button ( ), and the spectrum will zoom to its default view.
), and the spectrum will zoom to its default view.
Save Image: Click the Save Image button ( ), and then click in the spectrum.
), and then click in the spectrum.
Export Data: Click the Export button ( ) to save the (x,y) data points to a file.
) to save the (x,y) data points to a file.
Plot Axes: Click the Plot Settings button ( ). Specify the label and range for the x- and y-axes. Click OK and the spectrum will update itself.
). Specify the label and range for the x- and y-axes. Click OK and the spectrum will update itself.
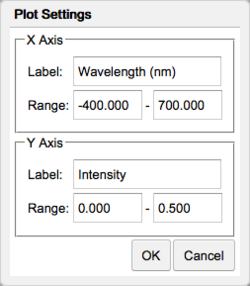
Plot Settings dialog