Login
To run WebMO, your system administrator will have established a WebMO user account for you and provided you with a web address, username, and password.
Start a web browser and type in the address of your WebMO implementation. The URL will be something like
- http://server.domain.edu/~webmo/cgi-bin/login.cgi

WebMO Login Page
Enter your username and password on the WebMO Login page and click the Login button. You will enter the Job Manager page.
Job Manager
After logging in, you will be on the WebMO Job Manager page. The left panel of this page displays your username and group, your job and total time limits, and the number of currently queued jobs. The top menu is for creating and managing jobs. The rest of the page lists the jobs you have submitted and/or run.

WebMO Job Manager Page
Click New Job: Create New Job in the top menu to start a new job.
Build Molecule
The Build Molecule page allows you to draw the 3-D chemical structure for which you wish to perform calculations. Initially the display in the page will be blank, indicating that no molecule has been drawn. The information panel on the left side of the page describes your progress in building the molecule, choosing a computational engine, setting job options, and submitting your job.
The 3-D editor has a menu bar on the top, toolbar on the left, and status line at the bottom. The menu bar (Build) and the status line (Build - C) indicate that you are currently using the build tool.

WebMO Build Molecule Page
The status line at the bottom of the editor window indicates the current tool and the editing operations that are possible.
Build the formyl fluoride molecule (HFCO):
- Click in the window to insert a C atom
- Select Build:O. Click near the C atom to insert an O atom. Drag between the two atoms to insert a single bond. Drag between the two atoms again to change the single bond to a double bond.
- Select Build:Other to bring up the period table, and click F. Click the C atom, drag away, and release to insert a single bond to a F atom.
- Select Cleanup:Comprehensive-Idealized to cleanup the structure. This idealizes all bond lengths and angles.
 1. C |
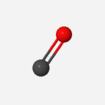 2. C=O |
 3. FC=O |
 4. HFC=O |
Optionally, choose Tools:View and View:Rotate, and drag the mouse in the editor window to rotate the molecule. View:Translate and View:Zoom work similarly. Observe the status line for the current tool and mode.
Optionally, choose Tools:Adjust. Click the O atom and then click the C atom to display the bond length in the status line. Click the F atom to display the bond angle.
Use the toolbar to accomplish common editing operations, including build ( ), periodic table (
), periodic table ( ), rotate (
), rotate ( ) and translate (
) and translate (![]() ), and adjust (
), and adjust ( ).
).
Click the forward arrow ( ) on the bottom right to continue.
) on the bottom right to continue.
Choose Computational Engine
The WebMO interface supports most popular computational chemistry programs including Gamess, Gaussian, Molpro, Mopac, NWChem, Orca, PC-Gamess, PQS, PSI, PWSCF, Q-Chem, Tinker, and VASP. These programs must be installed separately by the system administrator.
Choose which program to use. If the program is available on multiple computers, select the desired server.
NOTE: If only one computational chemistry program is installed, then this page is skipped.

WebMO Choose Computational Engine Page
Click the forward arrow () to continue.
Configure Job Options
The details of the Configure Job Options page depend on the specific computational engine chosen. Although reasonable defaults are provided, you may wish to:
- Enter a Job Name, i.e., an arbitrary name describing the calculation
- Specify the Calculation Type, e.g., Molecular Energy or Geometry Optimization
- Select a level of Theory, e.g., RHF
- Select a Basis Set, e.g., 6-31G(d)
You may also specify the overall Charge and Multiplicity of the molecule.
The Advanced Options tab gives you access to less commonly used options, e.g., symmetry, Cartesian coordinates, additional keywords, and number of processors (cores).

WebMO Configure Job Options Page

WebMO Advanced Job Options Page
Optionally, the Preview tab allows you to view and optionally edit the actual input file being submitted to the computational engine. Click the Generate button to view the input file.
Click the forward arrow () to submit your job. If the input file was previewed, click OK.
Job Manager (Revisited)
Jobs submitted to WebMO are queued and then run at the first available opportunity. If no other jobs are currently queued or running, then the submitted job will be run immediately.
The Job Manager page will display the status of your submitted jobs as: Queued, Running, Completed, or Failed. Click the Refresh menu item on the top of the page to update the status of your jobs.
The amount of CPU time spent on a job is displayed, so that you can monitor how long your job has been running. You may also view the output file of a running job by clicking the Raw Output icon ( ).
).
If necessary, you may terminate a queued or running job by clicking the Kill icon ( ). Jobs that exceed a user's compute time limits are automatically terminated.
). Jobs that exceed a user's compute time limits are automatically terminated.
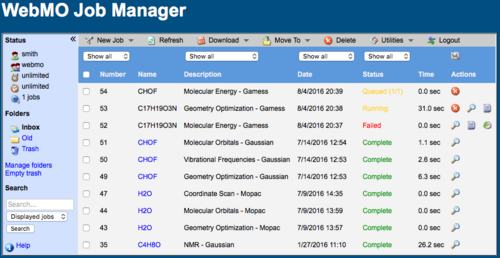
WebMO Job Manager Page with Queued, Running, Failed, and Complete Jobs
When the job is finished, the status will either be Completed or Failed, depending on whether the underlying program was able to complete the requested calculation. To view the results of a completed calculation, click the job name or  icon. If the calculation failed, click the icon to determine the reason for the failure, which is usually indicated near the end of the output file.
icon. If the calculation failed, click the icon to determine the reason for the failure, which is usually indicated near the end of the output file.
View Job
At the top of the View Job page is a display of the molecule's geometry, which can be rotated, translated, and zoomed using the View menu or toolbar buttons (, ![]() ,
,  ). To display a bond length, bond angle, or dihedral angle, click the select icon (), click the corrresponding atoms in order. Symmetry elements may be displayed using
). To display a bond length, bond angle, or dihedral angle, click the select icon (), click the corrresponding atoms in order. Symmetry elements may be displayed using  .
.
The remainder of the View Job page depends on the specific computational engine and calculation type. Information that can be displayed includes:
- Energy
- Symmetry
- Rotational Constants
- Dipole Moment
- Partial Charges
- Bond Orders
- Molecular Orbitals
- Vibrational Modes
- Excited States
- NMR Shifts
- Calculation Time
Clicking the view icon () can graphically display:
- Optimization Sequence Energies
- Dipole Moment
- Partial Charges
- Molecular Orbitals
- Normal Modes
- Infrared Spectrum
- Raman Spectrum
- UV-Vis spectrum
- NMR Spectrum
Clicking on the animation icon ( ) can display:
) can display:
- Optimization Sequence Geometries
- Normal Modes
- IRC Sequence Geometries
The complete output file of the computational chemistry program may be viewed by clicking the Raw Output link in the left panel.
The final geometry may be exported into a variety of format using the Export Molecule button.
A new job may be started using the final geometry of the current calculation by clicking the New Job Using This Geometry button.
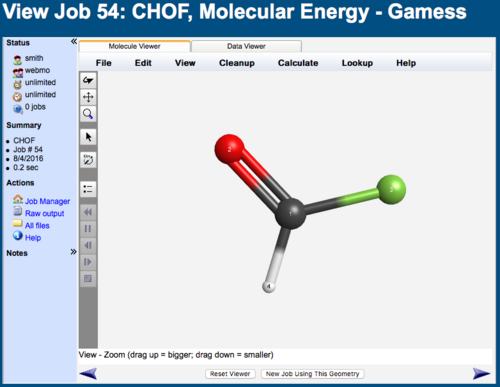
WebMO View Job Page (1/2)

WebMO View Job Page (2/2)