Molecular Orbitals
Electronic structure programs frequently have options to output various types of molecular orbitals.
The most commonly calculated orbitals are Canonical Orbitals (or "standard" molecular orbitals). Canonical molecular orbitals diagonalize the Fock matrix that defines the energy of the system, and the eigenvalues are therefore well-defined energies.
Any unitary transformation among the occupied Canonical Orbitals provides an equivalent overall wavefunction with the same total energy and electron density (wavefunction squared). Inclusion of some unoccupied orbitals in the transformation and using different criteria for the transformation definition can be useful for representing other quantities, such as bonding or electronic transitions.
Canonical orbitals are frequently delocalized across much of the entire molecule. Natural Bonding Orbitals are chosen to represent orbitals as two-center localized bonds and one-center lone pairs by using hybrid atomic orbitals and striving for two-electron occupancies. The result closely mimics Lewis structure theory.
Electronic transitions typically have contributions from multiple canonical orbitals, as can be seen in the raw output of an Excited State calculation. Natural Transition Orbitals are chosen to provide a compact representation of the electron and the hole resulting from the transition. The difference between the associated electron and hole densities approximates the change in electron density upon electronic excitation.
References:
Weinhold, Frank; Landis, Clark R. Discovering Chemistry With Natural Bond Orbitals. New Jersey: John Wiley & Sons. (2012)
Martin, Richard L. Natural transition orbitals. J. Chem. Phys. 118, 4775 (2003)
Natural Bonding Orbitals (NBO's)
NBO calculations are supported internally by Gaussian, Q-Chem, PQS, and TeraChem. The NBO program can also be obtained separately and externally linked to Gaussian, GAMESS, MolPro, and ORCA to provide NBO support.
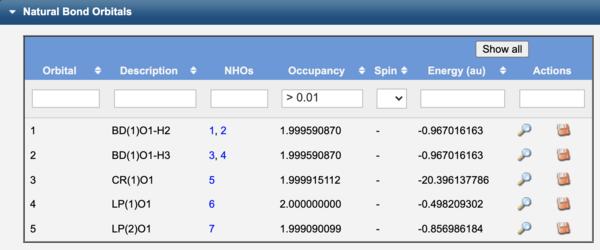
To compute the NBO's of a molecule, on the Configure Job Options page select a Calculation type of Natural Bond Orbitals. The View Results page will output the underlying Natural Atomic Orbitals (NAO's) that form Natural Hybrid Orbitals (NHO's), which in turn form the Natural Bond Orbitals (NBO's). These may be visualized using the corresponding View icon ( ).
).
NBO's of water
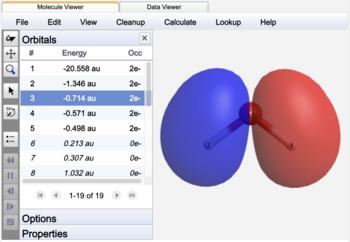 Molecular canonical orbital |
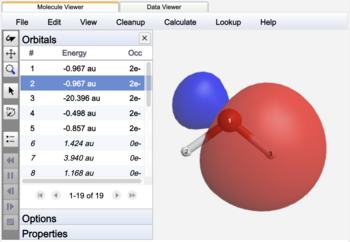 Localized natural bonding orbital |
The NBO's of water are label as bonds (BD), lone pairs (LP), or core (CR) orbitals. In contrast to Canonical orbitals that are delocalized across the entire molecule, NBO's are localized on atoms (CR, LP) or between atoms (BD) similar to Lewis structure electron pairs.
Natural Transition Orbitals (NTO's)
NTO calculations are supported by Gaussian and Q-Chem.
To compute the NTO's for an electronic transition, on the Configure Job Options page select a Calculation type of Electronic States and UV-VIS and select the Excited State Method. Then for Gaussian, on the Advanced tab in the Transition Density to State textbox, enter excited state number for the transition, eg, 1 for first excited state, 2 for second excited state, etc. This number will likely come from an earlier excited state calculation. For QChem, All NTO's are calculated.
The View Results page allows one to visualize the transition hole density (ρh) and the transition particle density (ρe) for an electronic transition. The difference between these (ρe - ρh) approximates the change in electron density for the electronic transition (purple = decreasing electron density, green = increasing electron density).
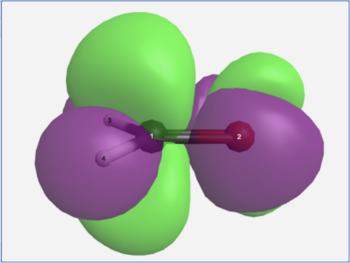 H2CO n→π* transition density difference |
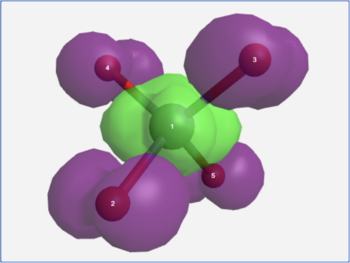 MnO4- charge transfer transition density difference |
It is important to recognize that purple and green in a density difference plot represent decreasing and increasing electron density. They are not the phases of a molecular orbital!
In the formaldehyde example, the S0→S1 electronic excitation causes electron density to leave the in-plane non-bonding orbital and enter the out-of-plane pi anti-bonding orbital. In the permanganate ion example, the ligand-to-metal charge transfer band in its visible spectrum is seen to arise from electron density leaving the outer oxygen atom lone pairs and moving into a d-orbital on the central metal atom.